

Inflammation and Aging
Can Endocannabinoids Help?
Aging often leads to cognitive decline due to neurodegenerative process in the brain. As people live longer, a growing concern exist linked to long-term, slowly debilitating diseases that have not yet found a cure, such as Alzheimer’s disease. Recently, the role of neuroinflammation has attracted attention due to its slow onset, chronic nature and its possible role in the development of many different neurodegenerative diseases. In the future, treatment of chronic neuroinflammation may help counteract aspects of neurodegenerative disease. Our recent studies have focused upon the endocannabinoid system for its unique effects on the expression of neuroinflammation. The basis for the manipulation of the endocannabinoid system in the brain in combination with existing treatments for Alzheimer’s disease will be discussed in this review.
Alzheimer’s disease (AD) is the most common neurodegenerative disease and accounts for the majority of diagnosed dementia after age 60. It is estimated to currently affect between 20 and 30 million people worldwide, with 4 million in the U.S. alone (Selkoe, 2005). The prevalence of the disease increases greatly after 60 years of age (1–3%) to reach around 30% of the population at 85 years old and up (Walsh and Selkoe, 2004). As life expectancy increases in developed countries, the incidence of AD, and its burden on healthcare, is very likely to increase dramatically in the next few decades. Two classes of drugs are currently used in the AD symptomatology: acetylcholinesterase inhibitors (Donopezil, Rivastigmine, Galantamine) and a single N-methyl-D-asparate (NMDA) receptor antagonist (Memantine). The first class of drugs tries to maintain brain levels of acetylcholine, known to decrease early on in AD, during the mild to moderate stages of the disease. The second category of drugs regulates post-synaptic calcium ion influx due to the action of glutamate at NMDA receptors and is recommended for use during the moderate to severe AD stages (Lleo et al., 2006, Parsons et al., 2007). Currently available drugs do not reverse or stop the progression of the disease but only relieve certain cognitive symptoms. Therapies currently under investigation either target amyloid protein (Schenk et al., 2005) or pro-inflammatory cytokines such as tumor necrosis factor-alpha (Tobinick and Gross, 2008). The complex nature of AD would advocate for the use of a multimodal drug approach that would also provide protection from the processes that underlie neurodegeneration. In the following paragraphs we will discuss the growing evidence supporting a role for the endocannabinoid system in the regulation of chronic brain inflammation associated with AD and the potential benefits to modulate that system in combination with memantine.
Endocannabinoids
Cannabinoid refers to naturally occurring or synthetic molecules mimicking the activity of plant-derived cannabinoids from Cannabis Sativa. Two types of cannabinoid receptors have been so far identified in the body (see Howlett, 2002 for review), named CB1 and CB2 (Matsuda et al., 1990, Munro et al., 1993). Discovery of cannabinoid receptors (CBr) lead to the finding of endogenous agonists for these receptors called endocannabinoids (EC). EC are derived from arachidonic acid, arachidonoylethanolamide (anandamide), and 2-arachidonoyl glycerol (2-AG), synthesized on-demand post-synaptically and released in response to the entry of calcium ions (see Di Marzo et al., 2005 for review). These EC in combination with the two known CBr constitute the endocannabinoid system (ECS). In the central nervous system (CNS), CB1 is overwhelmingly represented over CB2 and particularly abundant in cortical regions, the hippocampus, cerebellum and basal ganglia (Herkenham et al., 1991) while CB2 may be restricted to microglia (Nunez et al., 2004) or neurons in the brainstem (Van sickle et al., 2005) and cerebellum (Ashton et al., 2006). Deactivation of the EC is due to a rapid enzymatic degradation in the synaptic cleft or after membrane transport (Piomelli, 2003). The ECS is thought to be a neuromodulator (Vaughan and Christie, 2005) and an immunomodulator (Klein, 2005). In the CNS, the ECS can influence food intake, endocrine release, motor control, cognitive processes, emotions and perception. Cannabinoids treatment has been shown to be neuroprotective under many experimental conditions. Drugs that manipulate the ECS are currently evaluated in various diseases ranging from cancer to AIDS (Prentiss et al., 2004; Walsh et al., 2003) for their peripheral analgesic and immunosuppressive properties. Their anti-inflammatory actions may make them useful in the treatment of multiple sclerosis, Parkinson’s disease and AD (Maresz et al., 2005; Ramirez et al., 2005; Eljaschewitsch et al., 2006; Marchalant et al., 2007, 2008). Very little in vivo evidence to support the use of EC receptor agonists has been reported (Ramirez et al., 2005; Marchalant et al., 2007, 2008), although in vitro studies have found evidence for their anti-inflammatory effectiveness (Facchinetti et al., 2003; Ramirez et al., 2005; Sheng et al., 2005). Our recent work demonstrated the anti-inflammatory effect of a chronic treatment of a low dose of the CBr agonist WIN-55,212-2 (without psychoactive effects) on the consequences of chronic neuroinflammation induced by the infusion of LPS into the 4th ventricle of young rats (Marchalant et al., 2007). Moreover, that same anti-inflammatory effect was found using a non-psychoactive dose given by slow subcutaneous infusion of WIN-55,212-2 to healthy aged rats; these rats also demonstrated improved spatial memory (Marchalant et al., 2008). Our ongoing work in aged rats has shown that treatment with the CBr agonist WIN-55,212-2 increases neurogenesis in the hippocampus (Unpublished results). Our preliminary data suggest that the neurogenic and anti-inflammatory effects in aged rats are due to the agonist/antagonist properties of WIN-55,212-2 at multiple receptors (Unpublished results).
Neuroinflammation and Alzheimer’s disease
Microglial cells and macrophages are key elements of the brain inflammatory response during neurodegenerative diseases such as AD. Microglia’s morphology in its “resting” state is highly ramified and possesses a down-regulated phenotype (low or no detectable expression of cell-surface proteins such as MHC class I or II). Even during that so called “resting” state, those cells are highly involved in surveying their environment using their numerous cellular processes (Nimmerjahn et al., 2005). This resting state seems to be maintained by intercommunication between CD200 produced by neurons and CD200 receptor expressed on microglia (Hoek et al., 2000). We speculate that the loss of this equilibrium could lead to a chronic activation of glia such as seen during aging or associated with neurodegenerative diseases. Subtle changes in the brain homeostasis can then lead to a rapid morphological change and protein expression pattern of those microglia cells (to a pro- or anti-inflammatory response profile) depending on the nature of the brain injury and the stage of the inflammatory response (Stout et al., 2005). In the brains of patients with AD an atypical inflammatory response can be observed characterized by an activation of resident microglia possibly accompanied by monocyte infiltration from the blood (Akiyama et al, 2000). Those activated cells are often found surrounding amyloid plaques either due to the presence of amyloid itself or the presence of neurodegeneration.
Chronic neuroinflammation contributes to the pathophysiology of AD (Akiyama et al., 2000; Wenk et al., 2000). Long term use of anti-inflammatory drugs has been hypothesized to counteract with these processes and thus protect against the disease; epidemiological evidence supports the long term use of NSAIDs to reduce the prevalence of AD (Andersen et al., 1995; In’T Veld et al., 1998). Indeed, NSAID use for more than two years was found to be significantly associated with a reduced risk of AD (Breitner et al., 1994; In’T Veld et al., 1998). However, clinical trials so far have produced mostly negative results. Recent studies suggest that anti-inflammatory agents may have a preventative influence on the development of AD pathology even if they do not appear to slow progression of dementia (Breitner et al., 1994; Wenk et al., 2000).
Inflammatory processes are closely associated to the neuropathological and cognitive syndromes of AD (Akiyama et al., 2000). Post-mortem analysis of inflammatory markers (activated microglia cells) of AD patient’s samples of the entorhinal and frontal cortex clearly correlated with synaptic loss in those regions. That correlation between inflammation and cell loss was even greater than between cell loss and neurofibrillary tangle density (DiPatre and Gelman, 1997) or degree of deposition of amyloid (Terry et al., 1991). It is not surprising therefore that the cascade of immunological events that can be observed in the brain of an AD patient are found to occur very early in the progression of the disease in those same brain regions that later show the greatest concentration of senile plaques and atrophy (Cagnin et al., 2001). Moreover, the development of inflammation within neuronal populations and regions known to be vulnerable in the brains of AD coincide with the memory impairments observed in the early stages of AD pathology (Davis et al., 1999). The brain’s inflammatory response leads to a cascade of self-perpetuating cellular events including increased release of prostaglandins (Katsuura et al., 1989), enhanced release of glutamate (Emerit et al., 2004), and blockade of glutamate uptake by glia (Rothwell et al., 1997). Inflammation is also known to be able to relieve the NMDA channels from their magnesium ion blockade and increase nitric oxide levels, both inducing a dysregulation of calcium ion influx at the post-synaptic membrane. Any subsequent activation of NMDA receptors by glutamate may then enable a continuous entry of calcium ions into neurons, potentially overcoming the endogenous mechanisms regulating calcium ion homeostasis (Albin and Greenamyre, 1992; Chao and Hu, 1994). This unbalance in calcium influx can thus impair mitochondrial respiration, oxidative stress, as well as energy production and membrane depolarization (Emerit et al., 2004). The consequences of long-term, low level brain inflammation might therefore contribute to impairment of calcium homeostasis and alter its downstream signal-transduction cascades (Barry et al., 2005).
Endocannabinoids and Alzheimer’s disease: a useful tool in addition to existing medication?
As described in the previous paragraphs, there is strong evidence that inflammation contributes to the evolution of the AD pathology and that the ECS might be of some use in treating chronic inflammation observed in AD. More classically, AD is described mainly by two post-mortem histological diagnostic features that are extracellular amyloid deposition and Tau hyperphosphorylation forming intracellular neurofibrillary tangles (LaFerla et al., 2007; Walsh and Selkoe, 2007; Kuret et al., 2005). The amyloid plaques forming during AD are due to accumulation of non-soluble fragments of the amyloid protein in various region of the brain, notably the hippocampus, cortex and amygdala. These plaques are well described as part of the neurodegenerative process involved in AD (LaFerla and Oddo, 2005). Amyloid protein can also trigger inflammatory processes as well as long term changes in the NMDA receptor activity, and thus the control of calcium ion influx on the post-synaptic side, to negatively impact the function of those cells (Fiala et al., 2007; Wenk, 2006). Tau protein, a microtubule associated protein, when hyperphosphorylated can accumulate and form neurofibrillary tangles that in turn impair intra-neuronal communication (Ballatore et al., 2007). There is a growing amount of evidence of the possible implication of the ECS in the regulation of events occurring during the course of AD progression, particularly on the regulation of amyloid clearance and inflammation in vitro as well as in vivo (Campbell and Gowran, 2007; Benito et al., 2007). Post-mortem analysis of AD brains demonstrated an increased expression of CBr located on microglia within the amyloid rich plaques (Ramirez et al., 2005) although that seems to not be the case in other studies (Benito et al., 2003). An overall decrease of cortical CB1 receptor seems to occur during AD further away from the plaques (Ramirez et al., 2005); a finding that is similar to that seen in aged rats (Marchalant et al., 2008) with region-specific variability (Liu et al., 2003). Moreover, up-regulation of the fatty acid amide hydrolase occurs within plaques and might be responsible for increase in metabolites from anandamide degradation, such as arachidonic acid, and thus contribute to the inflammatory process seen in AD. CB1 receptor stimulation demonstrated interesting neuroprotective properties (Marsicano et al., 2003) and thus suggests that the ECS might influence neuronal survival and may offer protection from pathological process like amyloid protein in AD. Amyloid infusion in vivo is associated with gliosis and memory impairment; both effects were reversed by infusion of an inhibitor of EC reuptake (Van der Stelt, 2006). The use of an agonist of CBr, WIN-55,212-2, also proved to be effective in reversing memory impairment following i.c.v infusion of amyloid beta, possibly linked to prevention of activation of microglia (Ramirez et al., 2005). We’ve shown that WIN-55,212-2 can reduce the number of activated microglia produced by the long-term infusion of LPS into the 4th ventricle of young rats (Marchalant et al., 2007) as well as the naturally occurring microglia activation associated with normal aging in rats (Marchalant et al., 2008). Taken together the results of these studies using different experimental animal models strongly suggest a potential benefit of the ECS manipulation on the consequences and expression of brain inflammation associated with normal aging and AD. These results thus advocate for an investigation of the effects of ECS stimulation on the presence of brain inflammation in currently available transgenic mouse models of AD. Our work also suggests that if EC have an influence on chronic neuroinflammation in young or aged animals, those effects might not only be due to direct activity of the ECS on microglia cells but also on the demonstrated modulation of glutamatergic transmission by EC in the hippocampus (Haller et al., 2007; Nemeth et al., 2008). The indirect influence of EC receptor stimulation on inflammation through modulation of glutamate release has striking parallels with the action of the memantine, a drug currently approved for the treatment of symptoms in moderate to severe AD patients (Parsons et al., 2007).
Memantine is a low to moderate affinity antagonist of the NMDA receptor channel that is able to restore the glutamatergic homeostasis by reducing the signal to noise ratio in presence of excessive synaptic glutamate release in pathophysiological conditions (Parsons et al., 2007). Efficiency of memantine in the symptomatological treatment of AD has been confirmed by clinical studies and additional benefits seems to result from its combined use with cholinesterase inhibitors (Ditzler, 1991; Gortelmeyer and Erbler, 1992; Orgogozo et al., 2002; Wilcock et al., 2002; Reisberg et al., 2003; Tariot et al., 2004; Gauthier et al., 2005; Reisberg et al., 2006). Memantine’s mechanism of action can be explained by the fact that it is more potent and slightly less voltage-dependent than magnesium, and thus may serve as a more effective surrogate for magnesium ions (Parsons et al., 1993; Danysz and Parsons, 2003). Memantine can effectively block the tonic pathological activation of NMDA receptors induced by increased presence of glutamate and/or modification of the NMDA receptor activity by the presence of amyloid protein or chronic neuroinflammation (Miguel-Hidalgo, et al.,. 2002; Wenk et al.,2006).
Could there be any benefits to use both cannabinoid agonists and Memantine? Figure 1 posits a simple hypothesis that might explain how the combination of these two pharmacological agents could synergistically reduce brain inflammation in AD. The ability of cannabinoid agonist to reduce intracellular calcium entry (Mackie et Hille 1992) and also glutamate release (Wang, 2003) and thus avoid excessive calcium influx in the pre- and post-synaptic sides of the synapse could be beneficial, in addition to the regulatory function of memantine on the post-synaptic influx of calcium ions through the NMDA channel. The enhanced control over glutamatergic synaptic function might not be the only outcome of this combined treatment. We have previously demonstrated that both WIN-55,212-2 and memantine independently have anti-inflammatory properties in a model of chronic neuroinflammation (Rosi et al., 2006, Marchalant et al., 2007), suggesting that a dual therapy in AD may also be beneficial on the inflammatory side of the disease. Moreover, both ECr stimulation and memantine have a positive effect on neurogenesis (Jin et al., 2006; Aguado et al., 2005; Galve-Roperh et al., 2007 for review) that may contribute to normal hippocampal function.
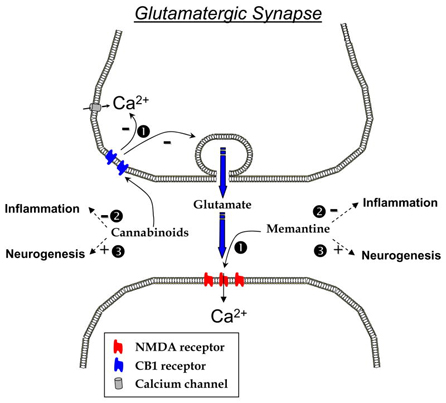
Overall, a multi-drug approach for AD seems to emerge as a potential alternative as none of the available drug therapies are capable of altering the progression of the disease. A multimodal treatment capable of limiting inflammatory processes may lead to a slowing of the disease progression, and may also reduce some of the cognitive symptoms and thus burden on families and care-givers.